{kind=link}
SpecView
SpecView is a graphical user interface (UGUI) program for the simulation and fitting of rotational, ro-vibrational, and ro-vibronic spectra. The application is written in Visual C++ and features MFC classes. A user-friendly environment allows for most spectra manipulation to be done with mouse moves and clicks. SpecView utilizes a DLL library that contains a large number of molecular spectroscopy models appropriate for the calculation of the energy levels and transition intensities of various molecules in different electronic states. The program includes a number of tools that allow the users to import experimental data, compare data to simulations graphically, and extract the simulated spectrum. It provides intelligent assignment of spectral lines and quantitative information on the energy level structure, spin-rotational wavefunctions, and transition intensities. For fitting molecular spectra, a Levenberg-Marquardt non-linear least-squares procedure is used. The program also supports fitting lineshapes of molecular spectra. The program has an intuitive environment and can be used for educational and demonstrational purposes since all molecular constants can be varied dynamically with spectra changes reflected immediately in the application's window.
One of the biggest advantages of SpecView is that the user can write their own spectroscopic model for specific applications. A sample model (coded in C++) has been provided in the Manual. Any user, even with minor knowledge in programming, should be able to extend the model library by creating their own *.dll file and registering it via the SpecView.ini file in the Windows system directory of the computer.
The SpecView program was developed in the research group of Prof. Terry A. Miller in the Spectroscopy Institute of the Ohio State University [1] and is now maintained by the research group of Prof. Jinjun Liu at the University of Louisville. For any questions regarding this program please email Dr. Jinjun LIu at j.liu@louisville.edu.
To install SpecView, please download the self-installing file SpecVi38a.exe at the bottom of this page. When it is downloaded and run on your local machine, it will produce a directory (unless told otherwise) C:\program files\OSU\specview\ which contains 3 folders: program, help and example. The program folder contains the executable SpecView.exe file. The help folder contains a manual in 3 formats: Word, html, and PDF. The example folder contains the source code for the various spectral models available in the first release.
The last released version of SpecView, 3.8a, has limited spectral simulating and fitting power. It contains only the most basic spectroscopic models for diatomic, linear, symmetric-top, and asymmetric-top molecules in unperturbed (Born-Oppenheimer) states. Newer versions of SpecView with more computing power and more spectroscopic models have been beta-tested extensively by the Miller and Liu groups. Especially, models for molecules in degenerate or nearly degenerate electronic states that are subject to vibronic interactions, e.g., the Renner-Teller (RT), Jahn-Teller (JT), and pseudo-Jahn-Teller (pJT) effects, have been developed by the Liu group.[2] A spectroscopic model for global simulation and fitting of ro-vibronic structure of JT and pJT molecules are under development. Second-generation SpecView program and advanced spectroscopic models are available upon reasonable request (email to: j.liu@louisville.edu).
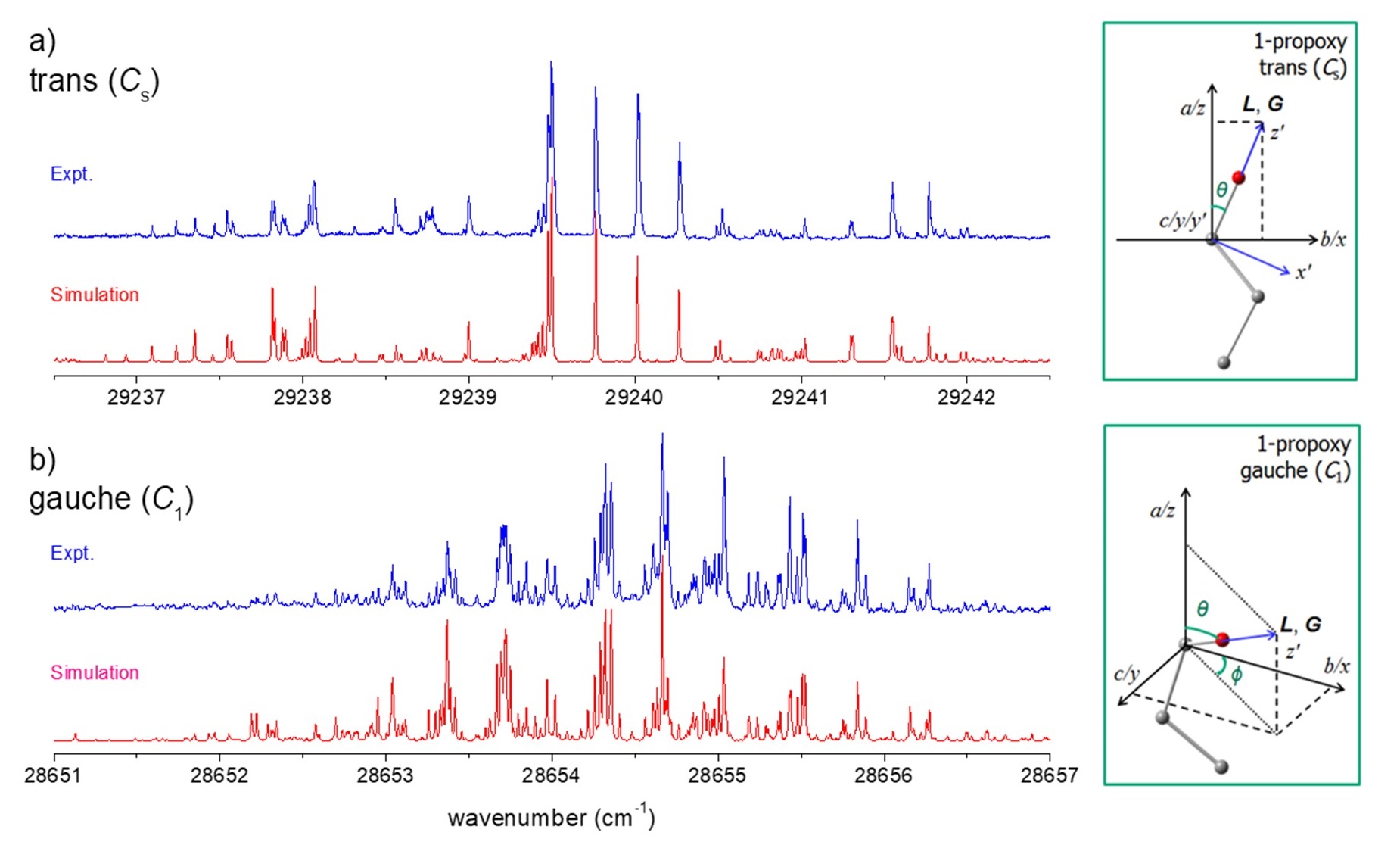
Figure. Experimental (blue) and simulated (red) spectra of the (a) trans- and (b) gauche-conformers of the 1-propoxy radical. Insets show the calculated molecular structures and the orientation of the orbital (L) and vibrational (G) angular momenta. (Hydrogen atoms are omitted for clarity.) The fitting and simulation was done using the SpecView program. Adapted from Ref. [2].
References:
[1] V. L. Stakhursky and T. A. Miller, “SPECVIEW: Simulation and fitting of rotational structure of electronic and vibronic bands,” in the 56th International Symposium on Molecular Spectroscopy (The Ohio State University, Columbus, OH, 2001), TC06,
[2] J.Liu, “Rotational and fine structure of open-shell molecules in nearly degenerate electronic states”, J. Chem. Phys. 148, 124112 (2018). DOI: https://doi.org/10.1063/1.5021946